
Abstract
BACKGROUND AND PURPOSE: Sotos syndrome is a rare autosomal dominant condition caused by pathogenic mutations in the NSD1 gene that presents with craniofacial dysmorphism, overgrowth, seizures, and neurodevelopmental delay. Macrocephaly, ventriculomegaly, and corpus callosal dysmorphism are typical neuroimaging features that have been described in the medical literature. The purpose of this study was to expand on the neuroimaging phenotype by detailed analysis of a large cohort of patients with genetically proved Sotos syndrome.
MATERIALS AND METHODS: This multicenter, multinational, retrospective observational cohort study systematically analyzed the clinical characteristics and neuroimaging features of 77 individuals with genetically diagnosed Sotos syndrome, via central consensus review with 3 pediatric neuroradiologists.
RESULTS: In addition to previously described features, malformations of cortical development were identified in most patients (95.0%), typically dysgyria (92.2%) and polymicrogyria (22.1%), varying in location and distribution. Incomplete rotation of the hippocampus was observed in 50.6% of patients and was associated with other imaging findings, in particular with dysgyria (100% versus 84.2%, P = .012).
CONCLUSIONS: Our findings show a link between the genetic-biochemical basis and the neuroimaging features and aid in better understanding the underlying clinical manifestations and possible treatment options. These findings have yet to be described to this extent and correspond with recent studies that show that NSD1 participates in brain development and has interactions with other known relevant genetic pathways.
ABBREVIATION:
- NSD1
- nuclear receptor binding SET domain protein 1
SUMMARY
PREVIOUS LITERATURE:
Sotos syndrome is a rare autosomal dominant condition with a well-described clinical manifestation, including craniofacial dysmorphism overgrowth, neurodevelopmental delay, and seizures. The genetic basis is a mutation in the NSD1 gene, which has a known part in neurodevelopment. Neuroimaging features previously described are macrocephaly, ventriculomegaly, and corpus callosum abnormalities. Few cases have been reported to have malformations of cortical development.
KEY FINDINGS:
This study aimed to provide a detailed neuroimaging phenotype of Sotos syndrome. The most intriguing finding in this study is the 95% prevalence of cortical development malformations, with dysgyria noted in 92.2% and polymicrogyria in 22.1%. Hippocampal incomplete rotation was observed in 50.6% and was associated with other imaging findings.
KNOWLEDGE ADVANCEMENT:
This study shines new light on the neuroimaging appearance of Sotos syndrome and adds to the classically described imaging stigmata some new and unique features including malformations of cortical development, hippocampal incomplete rotation, and anterior commissure hypoplasia. These findings support the presumed role of NSD1 in brain development.
Sotos syndrome is a rare autosomal dominant condition with an estimated prevalence of 1/14,000 live births,1 first described in 1964.2 The genetic basis of Sotos syndrome is caused by haploinsufficiency of the nuclear receptor binding SET domain protein 1 gene (NSD1), either through deletions or intragenic pathogenic variants.3,4 The nuclear receptor binding SET domain protein 1 (NSD1) protein is a methyltransferase that is known to play an important role in embryonic development.5
The main clinical features include craniofacial dysmorphism, overgrowth (macrocephaly, advanced bone age, and tall stature), and developmental delay and intellectual disability.6 Other features may also occur with varying frequency, including but not limited to scoliosis, conductive hearing loss, and cardiac abnormalities.1 Seizures have been reported in up to 25% of individuals with Sotos syndrome.7 Seizure semiology is diverse with febrile seizures, infantile spasms, absence, and tonic-clonic and myoclonic seizures.8 Few cases were reported to have drug-resistant intractable epilepsy.9
Neuroimaging findings in individuals with Sotos syndrome typically include macrocephaly, ventriculomegaly, dysgenesis of the corpus callosum,9 enlarged CSF spaces, prominent perivascular spaces, and abnormalities of the septum pellucidum.10 Despite the high prevalence of seizures, malformations of cortical development have been scarcely described but include heterotopias,11 polymicrogyria,12 and cortical dysplasias.13 Here, we aimed to expand on the neuroimaging phenotype by detailed analysis of a large cohort of patients with genetically confirmed Sotos syndrome.
MATERIALS AND METHODS
Study Design
This multicenter multinational, retrospective observational study aimed to perform deep phenotyping of neuroimaging findings in a large cohort of children with genetically confirmed Sotos syndrome. Site-specific institutional review board approval was obtained from all collaborating institutions before commencement, in accordance with the Declaration of Helsinki. Data are reported in line with the STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) statement (Online Supplemental Data).
Inclusion criteria were the following:
Genetically confirmed diagnosis of Sotos syndrome (ie, likely pathogenic or pathogenic variants in NSD1).
Sufficient clinical data available for interrogation
Good-quality, multisequence, multiplanar MRI of the brain, obtained by 1.5T or 3T scanners, with at least 3 sequences:
Sagittal T1WI
Axial T2WI or T1WI
Coronal T1WI or T2WI
Exclusion criteria were:
Individuals with pathogenic covariants in any other gene.
Clinical Data
The electronic health record was reviewed retrospectively by the referring clinician from each center and interrogated for demographic, clinical, and genetic information. Growth was assessed according to modified World Health Organization growth charts; overgrowth was defined as 2 SDs above the mean. Data regarding developmental delay (defined in children younger than <5 years of age) and intellectual disability (in children 5 years of age or older) were also collected. The seizure type was classified in accordance with the International League Against Epilepsy 2017 criteria.
Imaging Review
Brain MR imaging studies of each patient were reviewed by a central panel consisting of a pediatric neuroradiologist from the patient’s center and 3 other authors (K.M., S.S., and A.B.), all expert pediatric neuroradiologists (15, 20, and 10 years of experience, respectively). In case of disagreement among the radiologists, a joint review was sought to achieve a consensus. In patients with additional sequences or several imaging studies, all were reviewed, including follow-up MR imaging scans. The reviewing panel inspected the entire scan with particular attention paid to the cortex, brainstem, cerebellum, ventricular system morphology, and external dysmorphic features.
Malformations of cortical development, if present, were defined according to the latest published consensus classification,14,15 and the malformation subtype, distribution, and location were noted for each case. Dysgyria was noted when the cortex appeared to have variable thickness with a smooth gray-white transition and had an abnormal sulcation pattern, including sulcal depth irregularities, abnormal sulcal orientation, or excessive gyration (polygyria) not fulfilling the criteria of polymicrogyria. The hippocampal structure was noted as abnormal (incomplete rotation) if it presented as round or pyramidal or with a deep collateral sulcus.16
Statistical Analysis
Categoric variables were reported as frequencies and percentages. Age distribution was evaluated using a histogram. Because the age distribution was skewed, it was reported as median and interquartile range. The association between categoric variables was evaluated using the χ2 test or Fisher exact test, as appropriate. All statistical tests were 2-sided, and P < .05 was considered statistically significant. SPSS software was used for statistical analyses (SPSS Statistics, Version 29.0; IBM).
RESULTS
Seventy-seven patients met the inclusion criteria, of whom 48.1% were female, with a total of 114 brain MRIs. Age at the time of the first imaging study varied from 6 days to 16 years, with a median of 1.75 years (interquartile range, 4.9–0.8 years). One patient had a prenatal MR imaging that was reviewed as well as the postnatal scan.
All patients had a genetic diagnosis of Sotos syndrome, with a likely pathogenic variant or pathogenic variant in NSD1. Fifty-two patients had a full genetic report available, detailing 26.9% whole gene deletions, 25% missense, 26.9% frameshift (indel), 11.5% nonsense, and 7.7% splice site variants.
Clinical Presentation
Overgrowth was a core clinical feature in most patients, consisting of tall stature and weight gain in 59.7% and macrocephaly in 67.5% (Table 1). Developmental delay was noted in 88.3% of patients, and 58% of the patients were later diagnosed with intellectual disability of varying severity. A relatively high prevalence of autism spectrum disorder and attention deficit/hyperactivity disorder was found in our cohort, in 9 (11.7%) and 6 (7.8%) patients respectively.
Results: clinical features
Seizures were reported in 24 patients (31.2%), 9 of whom (11.7%) had abnormal electroencephalogram findings, with a variance in seizure presentation and response to medication. Thirteen patients had available information about the seizure subtype, of whom 8 (61.5%) had generalized seizures: 6 tonic-clonic, 1 myoclonic, and 1 combined myoclonic–tonic-clonic. Three patients (23.1%) had focal seizures, and 2 (15.4%) had febrile spasms.
Neurologic findings including hypotonia, nystagmus, and increased intracranial pressure were present in 71.4%. Ophthalmologic findings were present in 37.7%. Twenty-seven patients (35.1%) presented with idiopathic scoliosis (thoracic, lumbar, or combined) and were either observed only or treated with a back brace. Six patients (7.8%) presented with craniosynostosis. One patient was deceased at the time of writing secondary to complications of a pulmonary disease at the 2 years of age.
Neoplasia
Four patients (5.2%) presented with neoplasms, either malignant or benign. One patient presented with a tectal glioma and secondary obstructive hydrocephalus treated with an endoscopic third ventriculostomy. A second patient presented with an in utero adrenal mass, later diagnosed as a neuroblastoma in the perinatal period, which resolved spontaneously over the observation period. A third patient presented with a sacrococcygeal teratoma. The fourth patient presented with retinal melanocytoma, diagnosed at 1 year of age, showing spontaneous involution.
Imaging Findings
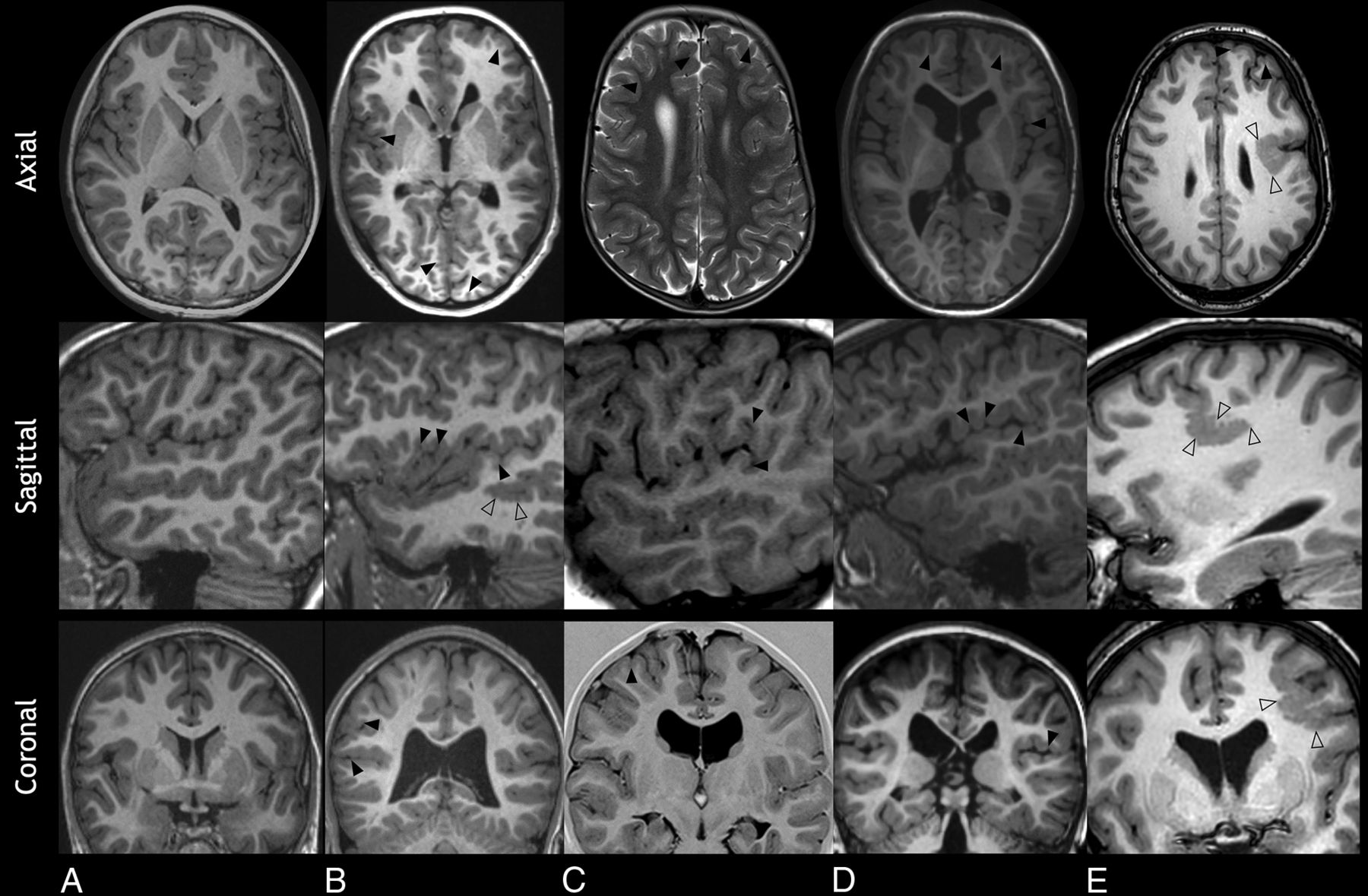
Malformations of cortical development were noted in 73 patients (94.8%) (Table 2). Most patients (71; 92.2%) exhibited dysgyria, which was typically bilateral and had a predominant frontal involvement in 59/71 (83.1%) and perisylvian involvement in 33/71 (45.2%). The main features of dysgyria were shallow sulci (42.3%) and polygyria (50.7%). Seventeen patients (22.1%) showed polymicrogyria, mostly bilateral, of which 12/17 (70.6%) were perisylvian (Fig 1). Most patients with polymicrogyria were noted to have dysgyria as well, with no significant statistical correlation (94.1% versus 91.7%, P > .999).

Malformations of cortical development in 4 patients with Sotos syndrome (B–E). A, An 11-year-old healthy control. Axial, sagittal, and coronal T1WIs show a normal appearance of the cortex and sulcation pattern. B, A 15-year-old boy. Axial and coronal T1WIs show diffuse dysgyria (arrowheads), and sagittal T1WI shows undulating gyri in keeping with polymicrogyria (empty arrowheads). C, A 5.5-year-old girl. Axial T2WI and coronal T1 inversion recovery show frontal dysgyria with shallow sulci in disorganized orientation (arrowheads). Sagittal T1WI shows perisylvian dysgyria. D, A 3-year-old boy. Axial and coronal T1WI shows asymmetric, left-sided, frontal dysgyria (arrowheads). Sagittal T1WI shows perisylvian dysgyria (arrowheads). E, A 13-year-old girl. Axial, sagittal, and coronal T1WI shows polymicrogyria with regionally increased gyral/sulcal frequency and greater corticomedullary junction irregularity compared with dysgyria (empty arrowheads), accompanied by frontal dysgyria (arrowheads).
Results: imaging findings
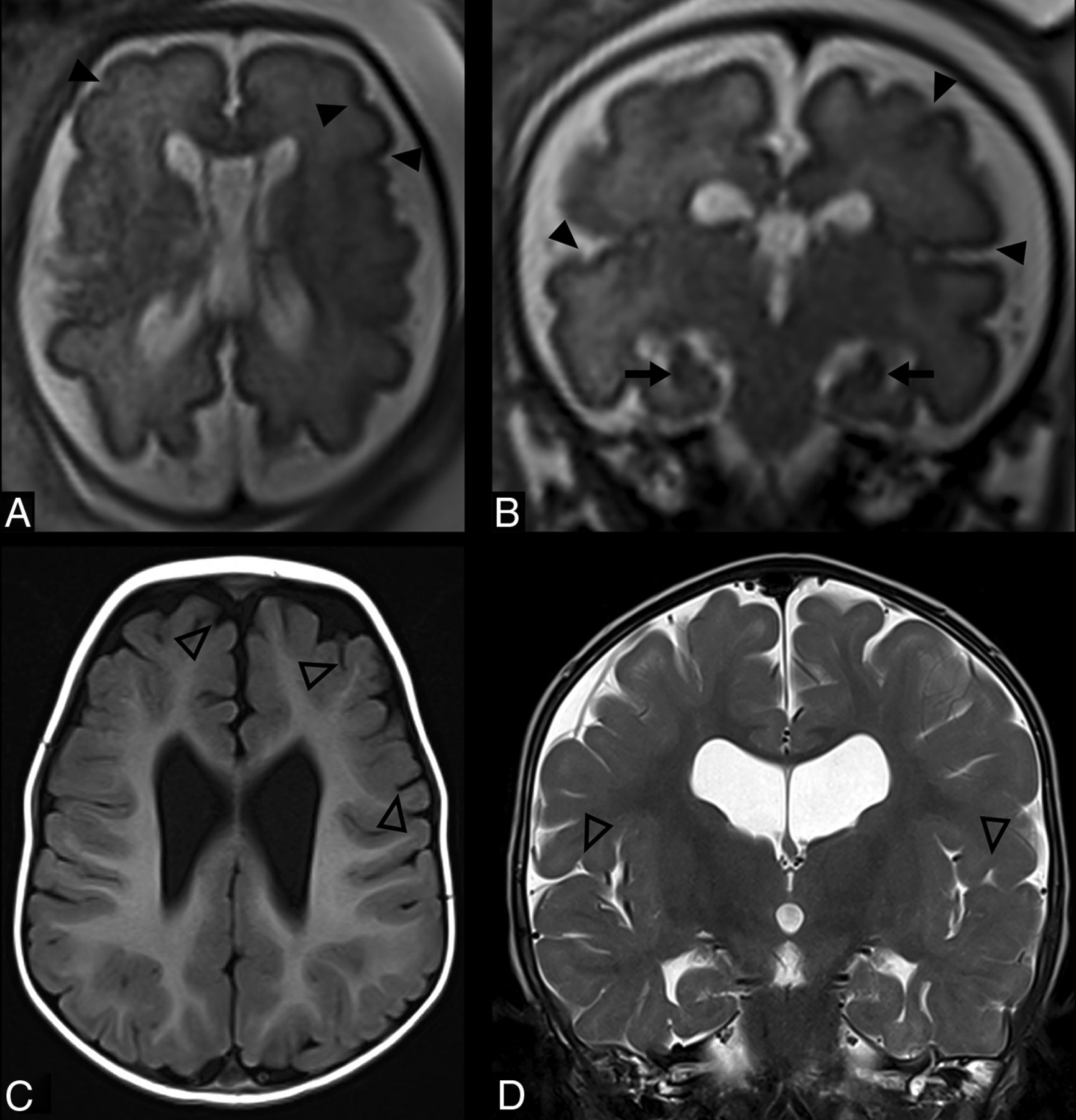
Incomplete rotation of the hippocampus was observed in 39 cases (50.6%) and was associated with other imaging findings (Table 3). Dysgyria was more common in patients with incomplete rotation of the hippocampus (100% versus 84.2%, P = .012) as well as a thin corpus callosum (79.5% versus 50%, P = .007) and white matter signal change (25.6% versus 2.6%, P = .004). The interthalamic adhesion (massa intermedia) was missing in 23.4%, and the anterior commissure appeared thin in 49.4% (Fig 2).

Imaging characteristics of Sotos syndrome in 3 patients (A, C, and D). A, A 16-year-old girl. Sagittal T1WI shows midline abnormalities including a thin corpus callosum (white dashed arrow), a thin anterior commissure (white empty arrow), and brainstem dysmorphism, a shallow pontomedullary sulcus (white empty arrowhead). B, A 16-year-old girl, an age-matched healthy control. Sagittal T1WI shows a normal midline appearance of the corpus callosum (white dashed arrow), anterior commissure (white arrow), and a pontomedullary sulcus (white arrowhead). C, A 10-month-old boy. Axial T2WI shows reduced white matter volume in the posterior cerebrum with enlargement of the ventricular atria and occipital horns and enlarged perivascular spaces (black arrows). D, A 3-year-old boy. Coronal T2WI demonstrates enlarged CSF spaces (asterisks), bilateral incomplete hippocampal rotation (black dashed arrows), and ventriculomegaly (black arrowheads).
Association of incomplete hippocampal rotation and other imaging findings
Structural anomalies were noted in most patients, including a macrocephalic skull (79.2%), ventriculomegaly (87.0%), corpus callosum abnormality (68.8%), ventricular dysmorphism (63.6%), and hypertelorism (58.4%). Less common were prominent perivascular spaces (39.0%), abnormalities of the septum pellucidum (33.8%), and multifocal patchy white matter signal changes (14.3%). In 23.4%, the brainstem was slightly dysmorphic, typically exhibiting a shallow pontomedullary sulcus, accompanied by cerebellar tonsillar ectopia in 18.2%.
No statistically significant correlations were found between imaging findings and clinical presentation. No association was found between seizure incidence and malformations of cortical development. Cortical malformations were seen in 91.7% of patients with seizures and in 96.2% of patients without seizures (P = .59), similarly for dysgyria (87.5% versus 94.3%, P = .37).
A single patient had a prior fetal MR imaging at 32 weeks of gestation due to macrocephaly on prenatal screening ultrasound and slightly elevated β-human chorionic gonadotropin. The scan showed biometrics consistent with macrocephaly as well as features of dilated and mildly dysmorphic ventricles, enlarged CSF spaces, hypertelorism, and incomplete rotation of the hippocampi. The cortical folding was appropriate for gestational age except for some asymmetry. This patient had a follow-up MR imaging at 1 year of age and showed mild ventriculomegaly, a thin corpus callosum, and mild dysgyria. (Fig 3).

A patient with Sotos syndrome. Fetal MR imaging at 32 weeks’ gestational age (A and B) and postnatal MR imaging at 1 year of age (C and D). A and B, Axial and coronal T2WI shows mild asymmetry of sulcation (arrowheads), mild ventriculomegaly, enlargement of the cavum septum pellucidum/vergae, and taller-than-wide hippocampal formations consistent with incomplete hippocampal rotation bilaterally (arrows). C and D, Axial T1WI and coronal T2WI show mild dysgyria in the frontal and perisylvian regions (empty arrowheads), ventriculomegaly, incomplete hippocampal rotation, and thinning of the corpus callosum. Distention of the cavum septum pellucidum/vergae has intervally resolved.
DISCUSSION
Imaging Findings
The most intriguing finding in this study is the 95% prevalence of cortical development malformations in patients with Sotos syndrome. This high prevalence has not been described in previous studies, most published before the term “dysgyria” was introduced. This finding may link the role of the NSD1 gene in brain development with the appearance on MR imaging.
Several previous studies have investigated brain development in Sotos syndrome, with only a few cases describing gray matter heterotopia11 and a single case report of polymicrogyria.12 There has been one additional report of an individual with polymicrogyria and an unbalanced translocation, including a deletion encompassing the NSD1 gene.10 The contribution of ≥1 of the numerous other deleted or duplicated genes to the imaging phenotype of this individual cannot be excluded, though our current findings are in support of the NSD1 deletion as a possible cause for polymicrogyria in that patient.
Incomplete hippocampal rotation can be a normal variant, especially when it occurs unilaterally on the left, or alternatively, the incomplete hippocampal rotation can be a developmental abnormality.17 In our cohort, incomplete hippocampal rotation tended to be bilateral and more common than in healthy children (50.6% of cases), suggesting that it may be secondary to regional temporal dysgyria from abnormal cortical folding. Normal hippocampal folding is theorized to be secondary to adjacent cortical development, thus potentially explaining concurrent cortical malformations.18 A statistically significant association was found between incomplete hippocampal rotation and dysgyria, suggesting that it may represent an imaging stigmata of Sotos syndrome.
Whitehead et al19 showed abnormalities involving the massa intermedia and anterior commissure to be more common in patients with midline abnormalities than in those without them. We found, in concordance, massa intermedia absence in 23.4% and a thin anterior commissure in 49.4% of patients with Sotos syndrome. These commonly affected midline structures should be evaluated as part of routine MR imaging reporting in patients with Sotos syndrome.
Slight dysmorphism of the brainstem, mainly a shallow pontomedullary sulcus, was observed in 23.4% of patients. It is most likely a feature secondary to the described cranial abnormalities; hence, no true malformation was observed, but it possibly represents another aspect of brain maldevelopment.
Other imaging findings corroborate previous reports, with macrocephaly, ventriculomegaly, and midline abnormalities observed in most MR imaging scans inspected and in all age groups.
Fetal Imaging
Published data regarding the prenatal presentation and imaging features of Sotos syndrome are sparse. Zhang et al20 described a case series of 7 patients with subtle nonspecific ultrasound findings such as ventriculomegaly. Lu et al21 published a case with fetal MR imaging showing ventriculomegaly and polyhydramnios. The findings on 1 fetal MR imaging included in our study are similar to those found postnatally for this child (Figs 2 and 3). However, the follow-up postnatal MR imaging revealed maturation of dysgyria, which was merely hinted at in the fetal MR imaging as asymmetry only. Together, cortical gyral abnormalities or asymmetries, abnormal hippocampal rotation, ventriculomegaly, and enlargement of the cavum septum pellucidum should raise the possibility of Sotos syndrome and the possible need for further genetic testing.
Neoplasia
Sotos syndrome, like other overgrowth syndromes, is known to entail an increased risk of malignancies.22 The prevalence of 5.2% in our cohort supports the evidence of an elevated neoplasm risk in these patients with a variance in tumor type and location.
Clinical Findings
The clinical findings in our cohort are consistent with the previous literature, with core features of overgrowth, psycho-cognitive impairment, seizures, scoliosis, and other neurologic or ophthalmologic abnormalities. The high prevalence of developmental delay and intellectual disability, autism spectrum disorder, and attention deficit/hyperactivity disorder have been described in previous studies.23,24 The somewhat lower prevalence of autism spectrum disorder and attention deficit/hyperactivity disorder may be explained by the young age in our cohort, patients not yet evaluated for such conditions. This understanding could enable early assessment and proper educational adjustments.
Genetic and Biochemical Literature Review
Several findings support the important role of the NSD1 gene in brain development and provide possible correlates for the observed malformations of cortical development in our cohort. Human expression data (Human Protein Atlas, Genotype-Tissue Expression Project [GTEx; https://www.genome.gov/Funded-Programs-Projects/Genotype-Tissue-Expression-Project]) demonstrate that NSD1 is expressed in the brain and in the cerebral cortex. It binds upstream of the promoter regions, thereby regulating transcription through interactions with methylated lysine 36 of histone H3 (H3K36) and RNA polymerase II.25 Oishi et al26 have shown that there is a predominantly neuronal expression of NSD1 in the cerebral cortex of mice. Complete knockout of NSD1 impaired migration and laminar positioning of cortical neurons27 and caused high apoptosis and an inability to complete gastrulation, ultimately resulting in lethality.5
A key downstream effector of NSD1 is the adenomatous polyposis coli 2 (APC2) gene. The APC2 gene has a crucial role in neuronal migration,28 and biallelic pathogenic variants in APC2 are known to lead to a neurodevelopmental disorder characterized by brain malformations, including cortical dysplasia and lissencephaly among others.29 Most interesting, Almuriekhi et al27 and Mastrangelo et al30 have described 3 individuals from 2 families with biallelic pathogenic variants in the APC2 gene, presenting with features resembling Sotos syndrome, including macrocephaly. Additionally, APC2 is a regulator of the Wnt signaling pathway, and there is substantial evidence of a connection between NSD1 and the Wnt signaling pathway,31 which could possibly contribute to the increased incidence of neoplasms in Sotos syndrome.
The findings of Quintero-Rivera et al32 suggest that NSD1 overexpression influences the PI3K/AKT/mTOR signaling pathway. Because the PI3K/AKT/mTOR pathway has a well-established role in cell growth, they propose that the undergrowth seen in NSD1 overexpression is related to the effect it has on this pathway. It would, therefore, be feasible that NSD1 loss of function may likewise cause overgrowth through the PI3K/AKT/mTOR pathway. Pathogenic variants in the genes of the PI3K/AKT/mTOR pathway are a well described cause of megalencephaly,33 once again linking NSD1 with malformations of cortical development.
Implications
The unexpected high prevalence of cortical malformations in our study carries important clinical-practical implications. When classic Sotos syndrome–related neurologic signs and symptoms are present, brain MR imaging should be performed and scrutinized for typical midline abnormalities and cortical malformations that may be subtle. The cortical malformations in Sotos syndrome often manifest as dysgyria or nonspecific cortical asymmetries on fetal MR imaging and, therefore, may be easily overlooked, especially without an optimal MR technique or when there are motion artifacts.
Limitations
Variability in imaging was an unavoidable consequence in this multicenter, retrospective study that included examinations acquired with various MR imaging hardware, magnet field strengths, and imaging protocols. Our inclusion criteria aimed to eliminate or at least diminish the effect of these differences. The clinical data were available in most patients and did not require patient re-examination. The multicenter structure does pose a strength in the reduction of selection bias. Furthermore, the wide consensus with an expert from each center reduces observer bias.
Brain malformation classification and terminology have had many revisions across the years, which may explain the previous underdiagnosis and under-reporting. Dysgyria, a rather recent term, was introduced in association with the Tubulin gene family pathogenic variants and has since been identified in other conditions. A qualitative assessment of dysgyria rather than a measurement-based grading may present a limitation of our study, and further studies should focus on such measurements of the cortical structure as well as better classifying dysgyria.
The assessment of the anterior commissure was only possible in patients with a high-resolution sagittal T1WI. Due to the differences in imaging protocols, we did not perform a measurement and used a binary qualitative criterion based on visual inspection. Further research regarding dysgyria subtypes, quantitative brain measurements, as well as genetic, histologic, and clinical correlations will be necessary for a better, repeatable, and precise classification.
CONCLUSIONS
Malformations of cortical development, anterior commissure hypoplasia, thalamic massa intermedia absence, and incomplete hippocampal rotation are common and under-recognized imaging features in Sotos syndrome, in addition to the classically described macrocephaly, ventriculomegaly, and callosal dysmorphology. These findings support the presumed role of NSD1 in brain development.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received March 21, 2024.
- Accepted after revision May 16, 2024.
- © 2024 by American Journal of Neuroradiology



{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.