
Graphical Abstract

Abstract
BACKGROUND AND PURPOSE: Duplication of the pituitary gland is a rare developmental anomaly. Multiple associated craniofacial malformations have previously been reported with the largest series to date consisting of 5 patients. In this multi-institutional series of 10 patients, we present a detailed review of the imaging features and discuss a possible overarching pathogenesis that would explain most of the detected malformations.
MATERIALS AND METHODS: Inclusion criteria for this retrospective imaging review were the presence of a pituitary stalk and gland duplication and the characteristic appearance of the hypothalamic ventral midline. In addition to the clinical presentation, we recorded the imaging findings of 10 patients (9 girls) through onsite and online reviews. Genetic analysis was available for 6 patients.
RESULTS: The duplicated pituitary stalk and gland showed normal imaging appearances in all patients. Mammillary bodies were clearly identified lateral to the characteristic prominence of the hypothalamic ventral midline. Strands of tissue extending to the anterior dura (“limited ventral myeloschisis”) were noted at the medulla oblongata in 10, and at the cervical spinal cord in 7 patients. The medulla oblongata showed a “butterfly” appearance on axial images in 9 patients. Ten patients had cervical segmentation anomalies (“zipperlike”), 9 had anterior-posterior brainstem patterning defects (small pons, elongated medulla), and corpus callosum measurements were abnormal in all patients. Three patients each presented with diencephalic-mesencephalic junction abnormalities and 4 with an anterior mesencephalic “cap.” An oropharyngeal teratoma was present in 4 patients. Genetics was normal in 3 of the 6 patients studied; the remainder were found to have mutations in EFNB1 and a gene variant of GIT1, 2 copies of 7 and 8 exon of SMN1 gene, and 2.126 megabase duplication at bands q11.1 and q11.2 of 1 chromosome 15, respectively.
CONCLUSIONS: Duplication of the pituitary gland presents as well-defined craniofacial and cervical spine malformation phenotype. Axial mesoderm duplication generating an excess of Sonic Hedgehog may be the primary embryologic driver leading to this condition.
ABBREVIATIONS:
- CFNS
- craniofrontonasal syndrome
- DPG
- duplication of the pituitary gland
- SHH
- Sonic Hedgehog
SUMMARY
PREVIOUS LITERATURE:
Previous reports analysing the imaging appearances in patients with DPG consisted mainly of single case reports. A characteristic association is the prominence of the ventral hypothalamic midline. Clinically, patients usually present with complications related to craniofacial dysraphism and presence of an oropharyngeal mass (ie, teratoma). Associated vascular and vertebral anomalies have been described. There is a strong female preponderance, the cause of which remains unclear. No unifying genetic cause for DPG has been identified. DPG needs to be differentiated from persisting embryonal infundibular recess.
KEY FINDINGS:
The prominence of the ventral hypothalamic midline in DPG represents excess midline tissue. All patients showed “limited ventral myeloschisis”, basilar artery and vertebral segmentation anomalies. Other frequent abnormalities were callosal dysgenesis, brainstem patterning defects, “butterfly” appearance of the medulla oblongata/attempted diastematomyelia, hypoplastic/absent olfactory bulbs, skull base defects and cleft lip/palate.
KNOWLEDGE ADVANCEMENT:
The term “mammillothalamic fusion” for the prominence of the ventral hypothalamic midline in DPG is discouraged. “Limited ventral myeloschisis” is described for the first time. The constellation of abnormalities associated with DPG suggest an axial mesoderm duplication with excessive SHH expression as the possible initial driver.
Duplication of the pituitary gland (DPG) is a rare developmental anomaly, often associated with multiple and complex craniofacial abnormalities that are thought to represent a defect arising early in development.1,2 Its cause has not yet been identified.
The complex but characteristic constellation of radiologic findings (sometimes called DPG-plus syndrome)1,2 may help in understanding the embryologic basis of this abnormality. Detailed knowledge of the imaging spectrum guides radiologists in their structured search for subtle anomalies that may otherwise be overlooked but can impact the patient’s management.
We present a series of 10 patients with DPG and describe in detail the spectrum of imaging abnormalities. We then discuss the clinical implications and propose possible embryologic mechanisms.
MATERIALS AND METHODS
Patients were retrospectively identified from medical records at 8 hospitals. Inclusion criteria were the presence of pituitary stalk and gland duplication as well as a characteristic appearance of the hypothalamic ventral midline (termed “prominent tuber cinereum” in previous reports) (Fig 1).3,4

Summary of imaging findings in pituitary gland duplication syndrome. A and E, Midline anomalies of the brainstem with characteristic prominence of the hypothalamic ventral midline (thick white arrows). Surplus tissue formation along the midline forms an anterior mesencephalic “cap” (arrowhead) and strands extending toward the anterior dura (“limited ventral myeloschisis,” white arrows and lower inset). There is an abnormal diencephalic-mesencephalic junction with incomplete separation of midbrain and tectal plate. B, Duplication of the pituitary gland and stalk with preserved posterior bright spot (black arrows). C, Cervical “zipperlike” segmentation anomalies. D, Skull base defect connecting the basal frontal bone to the maxilla (top) and attempted mandibular duplication (bottom). F, Top: Axial cut through the prominent hypothalamic ventral midline at the level of the mammillary bodies that are displaced laterally by the surplus tissue and separate from it. Bottom: “Butterfly appearance” of the medulla oblongata. G, Characteristic appearances of the short basilar artery. In addition, an aberrant artery arising from the ICA to supply the right anterior cerebral artery is noted. There is a network connecting the right and left anterior cerebral artery, but a typical anterior communicating artery is absent. H, Aberrant retropharyngeal arteries supplying the basilar artery (dashed arrows); MRA (right, patient 6) depicts the artery’s origin from the right subclavian artery, just distal to the common carotid and proximal to a rudimentary vertebral artery.
An initial imaging review confirmed the presence of pituitary stalk and gland duplication and prominence of the hypothalamic ventral midline. We identified several additional abnormalities that were subsequently assessed for all patients (presence or absence), namely limited ventral myeloschisis of the brainstem or cervical spinal cord, brainstem patterning defects (small pons, elongated medulla oblongata), anterior mesencephalic “cap,” thick lamina terminalis, diencephalic-mesencephalic junction abnormality, butterfly appearance of the medulla oblongata, callosal dysgenesis, intracranial lipoma, hippocampal malrotation, bilateral mesial temporal cleft, hypoplastic or absent olfactory bulbs, arterial anomalies, vertebral segmentation anomalies, accessory sutures, skull base defects (persistent craniopharyngeal canal, connection between basal frontal bone and maxilla), cleft palate, and midline oropharyngeal mass (Table).
Checklist for expected abnormalities in DPG
Reviews were conducted in consensus on site and via online meetings by all co-authors with first and last author (10 and 32 years of experience in pediatric neuroradiology) present for all reviews. All available data sets were reviewed (CT, MRI), including fetal MRI if available.
Measurements of the corpus callosum were obtained to better characterize its dysplastic appearances and compared with a previously published cohort of normal children.5
RESULTS
Patient Characteristics
Ten patients (9 girls) met the inclusion criteria. Patient characteristics are summarized in Supplemental Data. Nine patients presented for postnatal imaging due to concerns raised on antenatal ultrasound and/or fetal MRI regarding an oropharyngeal abnormality (cleft lip/palate, nasal fistula, or oropharyngeal mass) or brain abnormalities. Patient 3 presented for imaging with central precocious puberty.
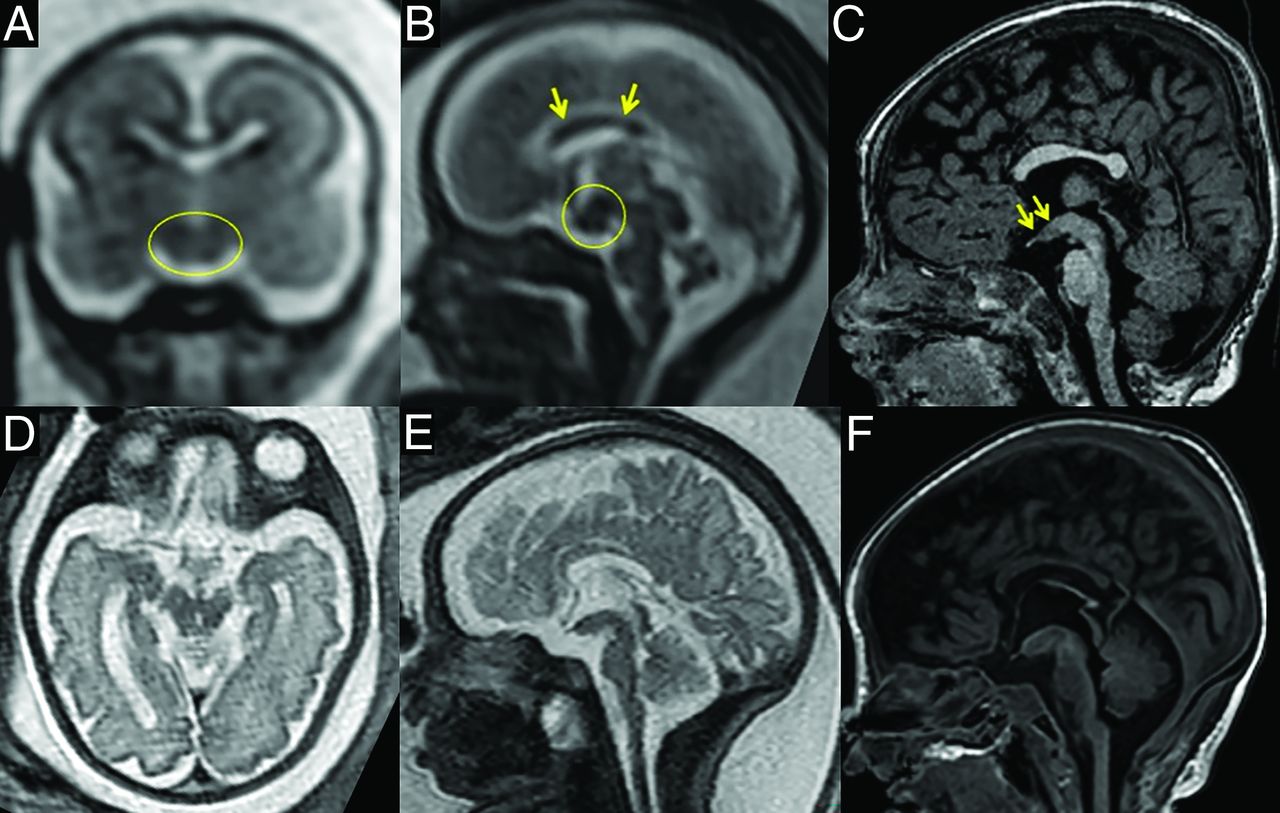
For this study, we reviewed postnatal scans only as they provided better image resolution, but fetal MRI, available for 2 patients, already showed the prominent hypothalamic ventral midline at 21 weeks gestational age (Fig 2).

Fetal MRI in comparison with postnatal findings. Thickening of the ventral hypothalamic midline is already present at 21 weeks gestational age in patient 5 (A–C, circles) and patient 4 (D–F).
Imaging Findings
MRI typically included a 3D volume data set (T1 or T2) and standard anatomic sequences (T2, T1, DWI) although this was variable and dependent on the clinical query for the scan. MRA was available for 4 and DTI for 3 patients. Head CT was available for 7, and spine CT for all patients. The imaging findings are summarized in Fig 1 and displayed in more detail in the Supplemental Data.
There was no focal abnormality of the 2 pituitary glands. The bright spot was present bilaterally in all cases. Notably, the mammillary bodies were clearly identified lateral to the prominent hypothalamic ventral midline in all cases, although intimately related to it in all patients (Supplemental Data). Patients 2, 3, and 5 were treated for central precocious puberty (Supplemental Data). Patient 5 was also diagnosed with borderline/partial adrenal insufficiency (low cortisol peak) at 8 years of age. Patient 6 had low cortisol levels initially, but normal synacthen test. Endocrine function was normal for patients 4 and 7–10. For patient 1 information on endocrine testing was not available.
The corpus callosum was dysplastic visually in 8 patients. Quantitative analysis of the corpus callosum compared with a reference cohort5 showed that the anteroposterior diameter was at or below the third centile in 9 of 10 patients (Supplemental Data). Measurements for patient 9 were normal apart from the length of the corpus callosum, which was below the third centile. In 1 patient no obvious dysgenesis was noted but the thickness of body and splenium was below the third centile. Another patient likely has an associated aberrant bundle based on callosal morphology; however, DTI was not available for confirmation.
An anterior-posterior brainstem patterning defect was present in 9 of 10 patients, characterized by small pons and long medulla oblongata on sagittal images. We also noted a “butterfly appearance” of the medulla oblongata on axial slices. DTI, available for 3 patients, revealed absent decussation of the superior cerebellar peduncles (patient 2), and an abnormal anterior-posterior midline pontine bundle (patients 5 and 8) (Supplemental Data). In addition, all patients showed anterior tethering bands, which we termed limited ventral myeloschisis, at the medulla oblongata; similar structures were noted along the anterior cervical spinal cord in 7 patients.
All patients had a malformation of the basilar artery which was short or nonfused (partial basilar artery duplication).6 In 2 patients (patients 3, 6), the basilar artery was supplied via a persistent artery coursing along the retropharyngeal space; MRA of patient 6 showed that this artery arose just distal to the right common carotid and continued along the retropharyngeal space to supply the basilar artery. MRA was not available for patient 3.
Eight of our patients presented with a skull base defect connecting the basal frontal bone to the maxilla. In addition, a persistent craniopharyngeal canal was noted in 7 of our patients. Seven patients presented with cleft palate and 4 patients with oropharyngeal teratoma.
Segmentation anomalies of the cervical spine were noted in 8 patients. The number of abnormal vertebral bodies in each patient varied, and in case of multiple levels involved, a “zipperlike” shape of midline separation was present.
We also identified the following abnormalities: abnormalities of the olfactory bulbs (7 patients), malrotation of the hippocampi (5 patients), bilateral mesial temporal cleft (4 patients),7 anterior mesencephalic “cap” (4 patients), diencephalic-mesencephalic junction abnormality (3 patients), and 1 patient each with thick lamina terminalis and bilateral accessory sutures of the parietal bones.
Genetic analysis was obtained in 6 patients, of whom 3 showed abnormalities, consisting of pathogenic variants in EFNB1 (patient 2), variants of unknown significance in GIT1 and SMN1 genes (patient 4), and a 15q11-q11 duplication of unclear clinical significance (patient 5).
DISCUSSION
Fewer than 60 cases of DPG have been described in the literature to date and a complete analysis of the radiologic findings based on a series of patients is lacking.8 We now contribute with a detailed analysis of 10 patients harboring a complex, but remarkably repetitive constellation of craniofacial abnormalities. Notably, all our patients presented with thickening of the ventral midline of the hypothalamus, which defines the DPG syndrome and was a criterion for inclusion in this study. DPG should be strongly suspected whenever this feature is noted, prompting assessment of the pituitary region.4 This malformation should not be mistaken for hypothalamic hamartoma, which presents in a different clinical context (often with gelastic seizures). In addition, mammillary bodies are often involved in hypothalamic hamartoma, while this is not the case in DPG. Axial images are useful to depict the lateral displacement of the mammillary bodies due to the prominence of the ventral hypothalamic midline in DPG. However, the most important findings that impact patient management are craniofacial dysraphism, the presence of an oropharyngeal mass (ie, teratoma), and associated vascular and vertebral anomalies.1 In particular, a short basilar artery and other vascular anomalies in association with a limited ventral myeloschisis of the medulla oblongata have not previously been described in such detail.
We noted a strong female preponderance in our cohort (1 boy among 10 patients) but also in the available literature.6,8,9 Of 52 reported patients, only 7 were boys, which in addition to our series compounds a striking 87.1% female prevalence. The cause of this phenomenon remains elusive, as does the genetic background to the DPG syndrome. One of our patients harbored EFNB1 abnormalities. Significantly, EFNB1 codes for the surface molecule EphrinB1, an important system for morphogenesis of the prechordal plate and notochord in zebrafish,10 and implicated in an X-linked dominant form of craniofrontonasal syndrome (CFNS) with greater severity in heterozygous females than in hemizygous males.11,12 In a recently reported series of CFNS, brain abnormalities such as dysplastic corpus callosum were reported, but no mention was made of pituitary gland abnormalities and the images were not available for review.13 The remaining genetic variants that were found have unclear clinical significance and do not seem to be associated with craniofacial syndromes.14⇓–16 We therefore surmise they were not pathogenic.
The radiologic appearances of DPG in our cohort differ from other malformations involving the third ventricle and pituitary region, for example, the persisting embryonal infundibular recess (Supplemental Data).17 On coronal images, there is apparent duplication of the pituitary stalk due to third ventricle floor extending into the pituitary stalk. Persisting embryonal infundibular recess may be associated with morning glory disc anomaly. Although this malformation is typically associated with a normal hypothalamic ventral midline, a recent report suggests that there may be a continuum.18 DPG also differs from other syndromic associations with vertebral anomalies, such as VACTERL or the oculo-auriculo-vertebral spectrum.19,20
Interpreting human malformation syndromes in terms of their embryologic origin is difficult because the phenotype is a result of the causal mechanism but also subsequent events in development. Furthermore, to the best of our knowledge, only 1 case of pituitary duplication has been studied neuropathologically.21 All other cases, including ours, have only been assessed by using neuroimaging, and therefore any assumption regarding the ultimate cause of this syndrome can only be retrospective. Still, some considerations can be advanced as working hypotheses awaiting confirmation from further experimental studies.
One of the salient features of these cases is the duplication of the pituitary gland (both, adeno- and neurohypophysis) and pituitary stalk. Furthermore, on coronal sections, the infundibulum is also duplicated.1,21,22 Thus, this malformation could be interpreted as a duplication of the most rostral ventral midline; in fact, the 2 duplicated lines are not located on a sagittal plane but a parasagittal position on either side of the midline. It has long been demonstrated, first in chickens and then in mammals, that the notochord plays a major role in the induction of the ventral midline of the neural tube.23 After transplantation of an additional notochord, the neural tube develops 2 floor plates (ie, ventral midline); between these lies a region containing motor neurons (basal plate of the neural tube). At the cranial extremity, the prechordal plate (the most cranial segment of the axial mesoderm) is mandatory for pituitary gland development.24 Furthermore, induction of an additional gland could be elicited by either axial mesoderm or ventral diencephalon.25 Finally, the secreted molecule Sonic Hedgehog (SHH), which is produced by both axial mesoderm and ventral midline of the neural tube, can control forebrain organization.26 All this evidence indicates that the rostral axial mesoderm plays a role similar to that of the notochord for more caudal levels. We therefore propose that duplication of the pituitary gland, pituitary stalk, and infundibulum could be secondary to prechordal plate duplication. Furthermore, such a duplication of the axial mesoderm will lead to an excess of SHH secretion.
The typical appearance of a prominent hypothalamic ventral midline has been variably interpreted as hypothalamic hamartoma,27 or thickened tuber cinereum by tubero-mammillary fusion.3,6 We disagree with these interpretations and propose an alternative model. By analogy with the results published previously,23 the midline tissue could correspond to the basal wall of the neural tube (ie, ventral hypothalamus). In 1983, Hori21 described the cells that are present in this tissue as magnocellular and medium-sized neurons associated with fibers; these large neurons are normally found in some of the hypothalamic nuclei. Unfortunately, Hori was unable to further characterize these cells since immunohistological techniques were unavailable at that time. However, his results support the theory that the thickened midline tissue is composed of supernumerary hypothalamic nuclei.
The presence of segmentation anomalies of the cervical spine, and particularly the zipperlike appearance in coronal planes, suggests that duplication of the axial mesoderm extends caudally to the cervical notochord. This feature reinforces our interpretation of axial mesoderm duplication. This is also the case for the apparent split spinal cord observed in patient 6 (Supplemental Data), further lending support to the hypothesis of SHH overexpression.
Teratoma (lying either in the oropharyngeal or nasopharyngeal space) was observed in several of our cases (patients 4, 6, and 7) and is also an inconstant finding in the DPG plus syndrome in the literature.2 Teratomas are thought to arise from embryonic stem cells first induced by proliferation and then differentiation. Interestingly, SHH is a promoter of embryonic stem cell proliferation.28 Consequently, an excess of SHH could be the trigger for teratoma initiation.
An anomaly of the basilar artery was found in all our cases. This vascular anomaly was first described in association with DPG by Shroff et al6 in 2003. In the mouse, Gli2 codes for GLI2, the main activator of SHH pathway. Gli2 -/- mouse lines, for which no GLI2 protein is produced, selectively lack the basilar artery,29 demonstrating the dependence of this vessel on the SHH pathway. Thus, complete, or partial duplication of the basilar artery could be a more likely explanation than a defective fusion. Furthermore, one can postulate that duplication of the basilar artery could result from notochord duplication or increased SHH signaling.
Duplication of the uvula and/or tongue, partial duplication of the mouth, or additional teeth were observed in 3 cases (patients 3, 4, and 7) and have been reported in DPG-plus syndrome.2 It is well known that SHH is mandatory for mouth opening and growth in both Xenopus and mouse.30 We propose that duplication of the oral structure could be associated with excess of SHH. The significance of palatal or orofacial clefts is more complex. Indeed, the presence of a tissue mass in the nasopharynx could prevent fusion of the palatal shelves as well as cause tongue duplication. However, it is interesting to note that an excess of SHH does generate such defects in the mouse embryo.31,32 Additional data are needed to understand the cause (or the causes) of such clefts.
Ciliopathies are associated with SHH defects and may aid in understanding some of the malformations observed in the DPG-plus syndrome. Ciliopathies represent a group of diverse diseases due to defective primary cilia, a cell organelle playing important roles in mechanoreception and in signal transduction. Notably, perturbation of cilia leads to SHH signaling defects with consequences on the ventral neural tube. By contrast, ciliary defects also impair proteasomal activity, relevant for the cleavage of GLI3 into its repressor form (GLI3R). Since GLI3R is a repressor of SHH signaling, the consequence will be an increase of SHH in the dorsal aspect of the neural tube. Defect of GLI3R processing has been proved as the cause of both olfactory bulb and corpus callosum anomalies in ciliopathies,33,34 and consequently an excess of SHH might also explain olfactory and callosal defects observed in our cases. The significance of the small anterior-to-posterior diameter of the corpus callosum, present in 9 of 10 patients analyzed, remains uncertain. Recently, it was shown that a short corpus callosum on prenatal imaging does not correlate with a specific genetic abnormality if present in isolation.35 However, in our patients, associated callosal dysgenesis was a frequent finding and related to a genetic abnormality.
Another interesting feature linking the DPG-plus syndrome with ciliopathy is the anterior mesencephalic cap dysplasia,36 or interpeduncular heterotopia.37 This abnormal tissue, lying in the peduncular fossa, is formed by aberrant nervous fibers,36 suggesting a defect of axonal guidance, and has been first described in Joubert syndrome, the main ciliopathy affecting the brain. Further anomalies have also been found in ciliopathies, such as absence of decussation of the superior cerebellar peduncles38 and abnormal pontine fibers.36 To the best of our knowledge, however, an abnormal anterior-posterior pontine bundle (Supplemental Data) has not yet been reported.
We also consistently observed a short clivus associated with a tissue stalk, or tethering band, inserted on the ventral surface of the pons or the medulla, which we termed limited ventral myeloschisis because it recalls the typical dorsal tethering bands found in limited dorsal myeloschisis. Such a defect was described by Manjila et al2 in 2012. and interpreted as a clival encephalocele, however without any pathologic proof. Significantly, however, already in 1983 Hori21 reported the same finding in the sole histopathologic description of a DPG case and found that the stalk was not composed of neural tissue but of connective and muscular tissues. The role of notochord on the development of the clivus ultimately remains obscure and needs further experiments in mammals.
The main limitation of our study is its small sample size, which may underestimate or overestimate the prevalence of the imaging findings described in our report.
CONCLUSIONS
We present radiologic phenotyping of the so far largest series of patients with DPG and associated craniofacial malformations. Our patients show a well-preserved, repetitive spectrum of ventral abnormalities involving the skull base, pituitary region, corpus callosum, tuber cinereum, brainstem, cerebral arteries, and cervical spine. Knowledge of this syndromic constellation can guide radiologists in the thorough interpretation of imaging studies in patients with DPG, thereby improving patient management. It is useful to include 3D T1WI of the brain (or CISS/FIESTA), cervical spine MRI, and MRA of head and neck and DTI. CT may be added to assess cervical segmentation anomalies. Endocrinology referral is recommended as patients may present with precocious or delayed puberty.39
The malformations associated with DPG suggest an axial mesoderm duplication with excessive SHH expression as the possible initial driver. Although SHH seems to be the most likely molecule involved in this process, we cannot rule out other factors that remain to be characterized.
Acknowledgments
The authors would like to thank all clinicians involved in our patients’ care.
Footnotes
Disclosure forms provided by the authors are available with the full text and PDF of this article at www.ajnr.org.
References
- Received July 13, 2024.
- Accepted after revision October 9, 2024.
- © 2025 by American Journal of Neuroradiology



{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.